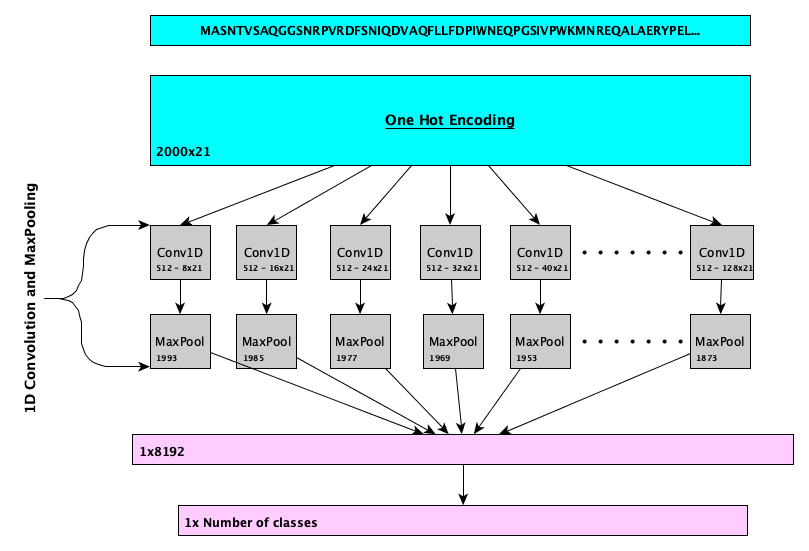
DeepGOWeb function prediction webserver
DeepGOWeb is a webserver for DeepGOPlus protein function prediction. It allows users to obtain predicted protein functions in three different ways. First, using the Prediction web page, users can submit protein sequences and obtain predictions which are downloadable in JSON format. Second, DeepGOWeb provides a REST API for users to access our servers programmatically. Finally, users can use our SPARQL endpoint to call DeepGOPlus within a SPARQL query.
Web page for submitting protein sequences
Users should provide the following data to use the service on the Prediction page:
- Format: FASTA format or Raw Sequences separated by a newline.
- Threshold: a value between 0.1 and 1.0 for filtering predictions by the confidence score of the model.
- Data: Protein sequences in the selected format. A maximum of 10 sequences are allowed in one request.
After submitting the request, users will be redirected to results page where they can see the predictions and download them in JSON format. Users can also save the link to the results page and come back to it anytime.
Output description
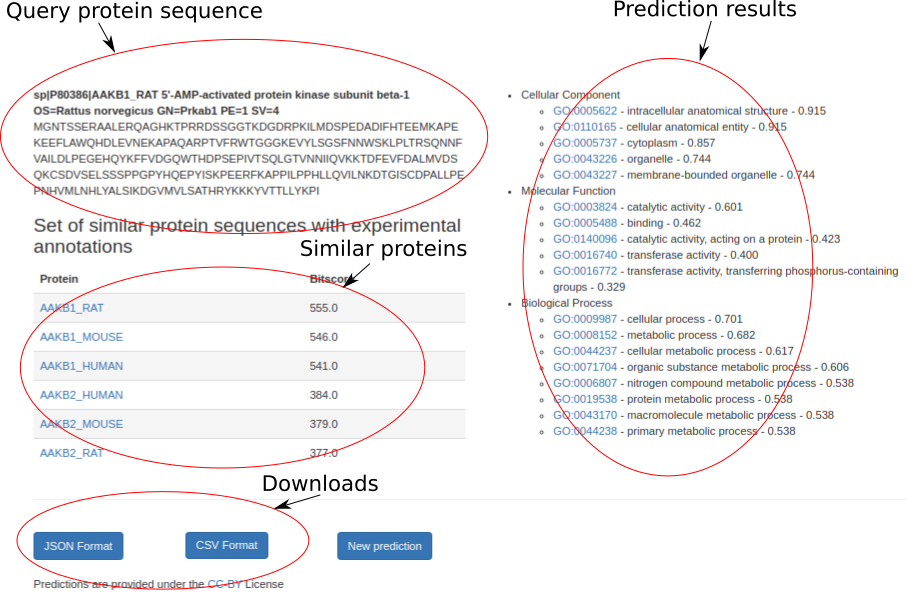
The output page provides function predictions for each protein. A prediction includes GO terms for the Biological Process, Molecular Function, and Cellular Component sub-ontologies with confidence scores. Only predictions with a confidence above the threshold parameter are shown. We also show a list of proteins that are used for predictions based on sequence similarity and their similarity score to the query protein. The results are provided under the CC-BY License and can be downloaded in JSON/CSV formats. Here is the example output page.
REST API
The REST API allows accessing the DeepGOWeb service programmatically. Here we provide an example using python and Requests library.
import requests
sequence = "MPYKLKKEKEPPKVAKCTAKPSSSGKDGGGENTEEAQPQPQPQPQPQAQSQPPSSNKRPSNSTPPPTQLSKIKYSGGPQIVKKERRQSSSRFNLSKNRELQKLPALKDSPTQEREELFIQKLRQCCVLFDFVSDPLSDLKFKEVKRAGLNEMVEYITHSRDVVTEAIYPEAVTMFSVNLFRTLPPSSNPTGAEFDPEEDEPTLEAAWPHLQLVYEFFLRFLESPDFQPNIAKKYIDQKFVLALLDLFDSEDPRERDFLKTILHRIYGKFLGLRAYIRRQINHIFYRFIYETEHHNGIAELLEILGSIINGFALPLKEEHKMFLIRVLLPLHKVKSLSVYHPQLAYCVVQFLEKESSLTEPVIVGLLKFWPKTHSPKEVMFLNELEEILDVIEPSEFSKVMEPLFRQLAKCVSSPHFQVAERALYYWNNEYIMSLISDNAARVLPIMFPALYRNSKSHWNKTIHGLIYNALKLFMEMNQKLFDDCTQQYKAEKQKGRFRMKEREEMWQKIEELARLNPQYPMFRAPPPLPPVYSMETETPTAEDIQLLKRTVETEAVQMLKDIKKEKVLLRRKSELPQDVYTIKALEAHKRAEEFLTASQEAL"
threshold = 0.3
r = requests.post('http://deepgoplus.bio2vec.net/deepgo/api/create', data={'data_format': 'enter', 'data': sequence, 'threshold': threshold})
result = r.json()
SPARQL
The SPARQL endpoint allows to call the DeepGOPlus function prediction model within a SPARQL query. We provide a custom SPARQL function called "deepgo" which takes a protein sequence and prediction threshold as an input and returns the predicted functions along with the subontology, label, and prediction score. The output can be downloaded in different formats such as json, xml, csv or text.
Example queries:
-
Example 1: Simple example query
PREFIX dg: <http://deepgoplus.bio2vec.net/functions#> PREFIX GO: <http://purl.obolibrary.org/obo/GO_> SELECT ?ont ?go ?label ?score { (?ont ?go ?label ?score) dg:deepgo("MPYKLKKEKEPPKVAKCTAKPSSSGKDGGGENTEEAQPQPQPQPQPQAQSQPPSSNKRPSNSTPPPTQLSKIKYSGGPQIVKKERRQSSSRFNLSKNRELQKLPALKDSPTQEREELFIQKLRQCCVLFDFVSDPLSDLKFKEVKRAGLNEMVEYITHSRDVVTEAIYPEAVTMFSVNLFRTLPPSSNPTGAEFDPEEDEPTLEAAWPHLQLVYEFFLRFLESPDFQPNIAKKYIDQKFVLALLDLFDSEDPRERDFLKTILHRIYGKFLGLRAYIRRQINHIFYRFIYETEHHNGIAELLEILGSIINGFALPLKEEHKMFLIRVLLPLHKVKSLSVYHPQLAYCVVQFLEKESSLTEPVIVGLLKFWPKTHSPKEVMFLNELEEILDVIEPSEFSKVMEPLFRQLAKCVSSPHFQVAERALYYWNNEYIMSLISDNAARVLPIMFPALYRNSKSHWNKTIHGLIYNALKLFMEMNQKLFDDCTQQYKAEKQKGRFRMKEREEMWQKIEELARLNPQYPMFRAPPPLPPVYSMETETPTAEDIQLLKRTVETEAVQMLKDIKKEKVLLRRKSELPQDVYTIKALEAHKRAEEFLTASQEAL" 0.3) . } - Example 2: Federated query which runs
DeepGOPlus on two sequences
from the UniProt SPARQL
Endpoint
PREFIX dg: <http://deepgoplus.bio2vec.net/functions#> PREFIX GO: <http://purl.obolibrary.org/obo/GO_> PREFIX rdfs: <http://www.w3.org/2000/01/rdf-schema#> PREFIX rdf: <http://www.w3.org/1999/02/22-rdf-syntax-ns#> PREFIX taxon: <http://purl.uniprot.org/taxonomy/> PREFIX up: <http://purl.uniprot.org/core/> SELECT ?protein ?organism ?isoform ?sub ?go ?label ?score WHERE { { SELECT DISTINCT ?protein ?organism ?isoform ?aa_sequence WHERE { SERVICE <http://sparql.uniprot.org/sparql> { ?protein a up:Protein . ?protein up:organism ?organism . ?organism rdfs:subClassOf taxon:9606 . ?protein up:sequence ?isoform . ?isoform rdf:value ?aa_sequence . } } LIMIT 2 } (?sub ?go ?label ?score) dg:deepgo(?aa_sequence 0.3) . }